aDiNA
about
Story behind the soil metagenome assembly paper
The Other Story Behind “Tackling soil diversity with the assembly of large, complex metagenomes”
This post accompanies our recent publication in collaboration with Janet Jansson, Stephanie Malfatti, Susannah Tringe, Jim Tiede, and Titus Brown. The paper is openly accessible on the PNAS website. There’s also been some well-written press releases that summarise the effort, and my mom told me she read these rather than the paper :) Additionally, Titus has written up a really nice blogpost about our story behind the paper, mainly from a technical perspective. This blogpost is an extension of that story from my (the postdoc’s) personal experience, and what it felt like to deal with this “Grand Challenge” project.
Summary of the paper
In short, the paper describes our solution to asking “What sequences is this read biologically connected to?” when you’re comparing it to billions to trillions of sequences. The solution lies in the ability to store and query the data cleverly, allowing us to both compress and separate essentially genomes from challenging metagenomic datasets. The effort published is largely the “proof” that these methods work on complex datasets using the HMP mock community, specifically for the purposes of assembling genomes from metagenomes. We used these efforts to assemble two major soil metagenomic efforts, revealing how even with concerted efforts we can only capture the “tip of the iceberg” of soil biodiversity and that we know essentially nothing about much of what we can access. Soil is a really challenging system! The paper was reviewed by both experts in the field of soil microbial ecology and sequencing assembly, and I think this resulted in a manuscript that can be interesting and useful to a wide audience. A challenge for me was making this happen in the six page constraint of PNAS – assemblers wanted more ecology and ecologists wanted more explanations about the bioinformatics. I hope the paper is useful, it complements a whole suite of tutorials to make these approaches more accessible. Here, I wanted to share my experiences in this long endeavor.
My story behind the paper
I joined the teams of Tiedje and Brown in 2009, having completed my PhD at the University of Iowa. At that time, I came in well-trained on what I would call “traditional microbiology approaches” – culturing, isolate characterization, biochemical activity assays – as well as having dabbled in proteomic and metaproteomic approaches. A challenge to my PhD was that I always felt like we were working with an ideal model system which represented only a “speck” of the community in the “environment”.
This frustration accompanied my curiosity towards the birth of metagenomics and the advances of high throughput sequencing. Fortunately, the NSF recognized the need to offer incentives for training in this field, and Jim Tiedje – microbial ecologist extraordinaire – was willing to write that proposal with me and even found funding for me in case things fell through. Five years later, Jim admitted to me that he welcomed me into his lab because I was an engineer and “engineers know how to program”. Not too often is Jim tricked…and I would argue that the majority of engineers don’t know how to program, with the exception of computer/electrical engineers.
I naturally entered this postdoc with excitement but soon found the first two years to be really, really depressing. Rather than being the expert I expected to be from my PhD, I was completely lost, struggling with the most menial programming tasks, from parsing files to installing programs. Reflecting back on the state of affairs at the time, I’ll refer you to the two figures below to give you some perspective on the problems I was being asked to solve while essentially learning to code.
This first figure shows the rapid growth of NGS technologies, outpacing Moore’s law. I entered in 2009, right when the red sequencing line starts growing faster than blue lined computational resources. For the record, DON’T DO THIS AS A POSTDOC. Moving from benchwork to computational efforts, this meant that tools I was working really hard to learn how to use were essentially not usable and resulted in lots of banging my head against the wall…and then some innovation fortunately.
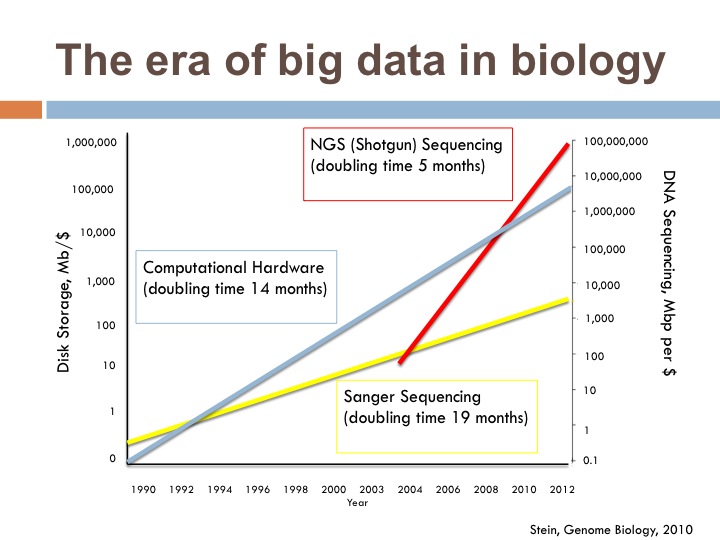
This next figure re-emphasizes the particular challenge I was facing. Not only did the challenge happen and sustain, it got worse. Within the first two years of my postdoc, the amount of sequencing I was used to multiplied by over a million fold. Yeah…

And these were the two years I was supposed to be super productive, and the timeline that many employers look at critically for any job I apply to (See Titus’s awesome post on how folks don’t know how to evaluate this). I admit that I was unable to produce much during this time. And I’ve only now come to terms with that being just fine with me. With a great team, particularly Titus, Jason Pell, Likit Preeyano, and Qingpeng Zhang, I learned about probabilistic data structures and got answers to my stupid programming questions. Jim and Titus never once asked me in these two years why I wasn’t writing and never once did I feel like they doubted me or the value this work (honest truth). I never heard any complaints about the pace; in fact, it was the opposite, these guys had my back at every meeting these concerns were expressed. I met Greg Wilson of Software Carpentry who patiently watched me fail teaching computer programming from a textbook and taught me to teach from my own experiences.
The idea to benchmark methods on the HMP mock metagenome came from my need to really understand the effects of our approaches with some sort of “groundtruth,” an attitude that likely grew from my naive experience in the field. Titus actually resisted this effort for a little bit, complaining legitimately that real metagenomes are so much more complex than a mock community. In the end, this effort really sold us on its application. The paper as I said attempts to demonstrate the robustness of these methods to the wide diversity of investigators that could use it, and my background in microbiology and my growth into receiving “a genuine bioinformatician” medal (mentally) really helped me write the text…which I hope is approachable to all.
Take home career message
Titus ends his blog on this effort with a take home career message stating that he’s not sure if he’d have taken on this project if he’d understood the problem better and admits that he has had challenges getting funding for environmental metagenome assembly. From my perspective, I don’t think I would’ve done anything different, and I really hope leaders like Jim and Titus evaluate that. I was ok doing a long postdoc – I wasn’t sure what I wanted to do at the time, and this adventure really helped me find my direction. I love working on complex systems, and it took quite a bit effort but it also built a lot of character. I find myself very comfortable defending my efforts and am getting rewarded now with progress on many projects that this effort enabled. With Kirsten Hofmockel at Iowa State, I have received an exciting grant – not to work on metagenomic assembly, per se – but to work on the metagenomic analysis of soil omic-approaches. This victory was huge for me. Partly because it strengthens my CV which is very slim on publications (currently), but also because I think this supports the investment (in so many ways) that both Titus and Jim put into me and the opportunities of the research. In my junior career, I’m witnessing others start to understand the challenges we’ve faced and expectations are shifting as well.
I’m continuing to build upon many of the computational approaches I’ve worked on, some with Titus and some on my own. I’ve applied these methods to viruses in the gut microbiome and to drivers of carbon metabolism in the soil. From a PhD-holding environmental engineer with very little hopes of finding a successful career, things look very optimistic on my end. For researchers who are making this transition now, embrace the change and give yourself time to do it. Be flexible. It’s an awesome ride to love your research and see it change lives.
blog comments powered by Disqus